BACKGROUND
MRKH syndrome is a congenital anomaly characterized by an aplastic or hypoplastic uterus, as well as an absence of the cervix and the upper third of the vagina,1 affecting approximately 1 in 5,000 women.2 It is the second most common cause of primary amenorrhea.3 Consequently, the diagnosis is usually made around puberty.4
As part of Müllerian duct agenesis, the phenotypic expression of MRKH syndrome may be limited to the reproductive tract (MRKH type 1), or in association with extragenital tract anomalies, usually of renal and skeletal origin (MRKH type 2). The presence of a pattern of multiple anomalies has been described, specifically characterized by aplasia of the Müllerian ducts (MU), renal aplasia (R), and alterations of the cervicothoracic somite (CS), which has been called the MURCS association (an acronym for its acronym in English).5 It occurs between 10% and 32% of cases and is classified as the most serious form of presentation of MRKH type 2.6
Patients with MRKH syndrome develop functional ovaries, complete external genitalia, and regular secondary sexual characteristics.7 They have a karyotype of 46,XX, and typical female development of external genitalia and secondary sexual characteristics. Despite the generally normal development and function of the ovaries, women with MRKH syndrome often present with primary amenorrhea.8 Clinical examination usually reveals a normal female phenotype with breast development, axillary and pubic hair, and normal external genitalia.9
In this article we present the case of a patient who was initially classified as having MRKH syndrome; however, due to the particular findings found within the approach, it was concluded that she presented an extremely rare congenital anomaly. The objective of this review is to evaluate the literature available in electronic databases, including case series, incidence reports and diagnostic approach, with the aim of expanding knowledge regarding this condition.
CLINICAL CASE
A 34-year-old patient, who attended the Gynecology evaluation for the first time, reported primary amenorrhea, as well as the recent onset of active sexual life with limitation in penetration and dyspareunia. Relevant history included a premature, natural birth with low birth weight and low height for the gestational age. Additionally, the patient was breastfed.
She presented slow normal pondo-stature psychomotor development compared to her siblings. She reported apocrine activity at 13 years of age, telarca-adrenarca-pubarch between 13-14 years of age, and very good interpersonal relationships and school achievement. She denied medical, chronic or degenerative diseases. She denied allergies, history of transfusions, chronic consumption of medications, drug addictions as well as surgical interventions. Additionally, she reported first and second-degree relatives without significant medical diagnosis.
The physical examination found that the patient weighed 63 kg, height 1.40 m (father 1.70 m and mother 1.65 m), and vital signs within normal parameters. On general inspection, there were no characteristic facies and no craniofacial alterations. However, the head was not centered to the rest of the body and the left scapula presented lower than the right. She had shoulder asymmetry, a shortened thorax, and an Adams sign with a shortened right hemithorax. She had obvious spine curvature and developed anatomically normal breasts and axillary hair. There were no abnormalities on static and dynamic inspection
Exploration of external genitalia, according to age and sex, found pubic hair present and normally distributed, a hyperpigmented labia majora that was anatomically normal. Vaginal examination found a shortening in length, approximately 3 cm, with no palpable cervix; the bimanual examination without delimiting the uterus or adnexa. Symmetrical extremities were without distal neurovascular compromise. An endocavitary ultrasound was performed in which no uterus was observed; however, we did observe images that suggest ovaries.
According to the protocol, general laboratory studies, thyroid profile and gynecological profile were requested to assess endocrine function. Additionally, we requested office studies based on the pelvic ultrasound, computed tomography (CT) and peripheral blood karyotype. An appointment was made with a report of results for evaluation. She subsequently attended with laboratory studies shown in TABLE 1. Diagnoses were integrated due to findings of hypothyroidism and hyperprolactinemia. In the study, treatment was initiated by the endocrinology service based on Levothyroxine 50 mcg per day, supported by the fact that in hypothyroidism, due to the decrease in thyroid hormones increases the levels of thyrotropin-releasing hormone (TRH), generating an increase in thyroid-stimulating hormone (TSH). Additionally, hyperplasia of lactotroph cells occurs and as a consequence hyperprolactinemia10; this is due to the stimulatory effect of TRH on lactotroph cells, or due to a reduction in hypothalamic dopamine content secondary to compression of the pituitary stalk.11
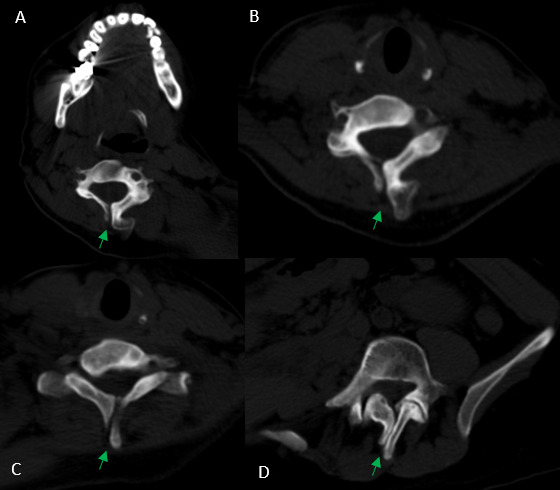
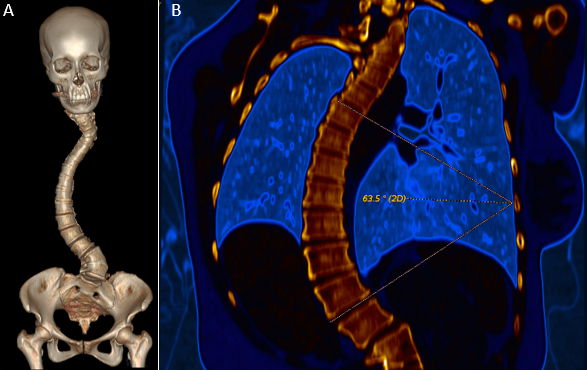
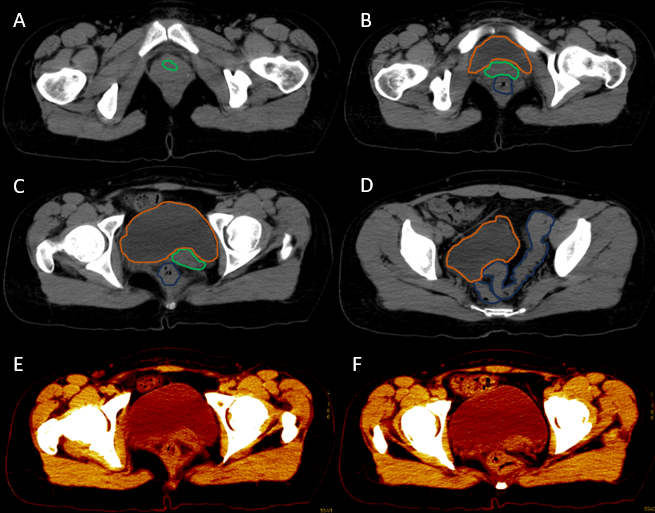
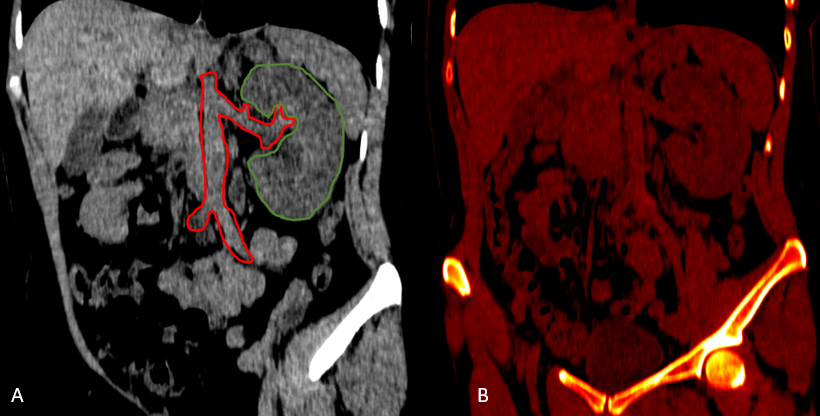
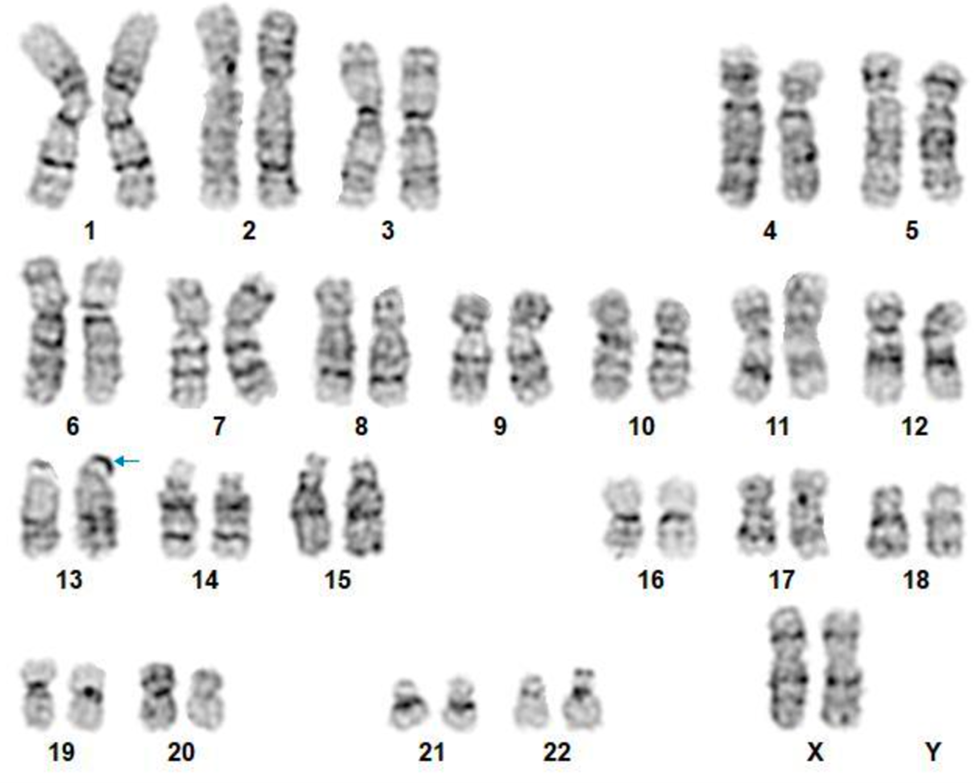
After analyzing the tomographic reports, the findings described in each of them were found in the cervical vertebrae (IMAGE 1), vertebral column (IMAGE 2), and abdomen (IMAGES 3 and 4). Karyotype report described in IMAGE 5.
_and_lumbar_(d)_vertebrae__there_are_.png)
_volumetric_reconstruction_of_the_axial_skeleton_b)_computed_tomography_in_coronal_sectio.png)

**_there_is_right_renal_agenesis__t.png)

With the above, diagnoses of: MURCS association (uterine, tubal, cervical and upper two-thirds of the vagina aplasia, renal aplasia and spinal dysraphism in cervical and lumbar vertebrae, as well as scoliosis), hypothyroidism and hyperprolactinemia were integrated. The patient was questioned about her reproductive desire; however, she did not want it. She was sent to an outpatient spine clinic for evaluation, and to nephrology for close and educational follow-up. She was scheduled for an appointment in three months with a new hormonal profile, for possible modification of the established treatment, and to consider starting a dopamine agonist.
MATERIALS AND METHODS
A search was performed in the Medline database via PubMed using the following terms: “Mayer–Rokitansky–Küster–Hauser Syndrome”, “müllerian anomalies”, “genetics of agenesis/hypoplasia of the uterus and vagina”, “uterine cervical aplasia and agenesis”. The search was limited by the following filters: “Case Reports”, “Review”, “Systematic Reviews”, and “Books and Documents”, “Spanish and English”, from 1979 to 2023, in total 316 studies were found. Editorials, reviews, and duplicate articles were excluded, mainly due to their low level of trust and veracity. 58 studies were selected that included women with a diagnosis of MRKH or MURCS association.
DISCUSSION
Among the most frequent causes of primary amenorrhea are gonadal dysgenesis and Müllerian aplasia, the latter can occur in isolation or be accompanied by abnormalities in various organs and systems.12 A severe form of MRKH type 2 syndrome is the MURCS association, which It is characterized by abnormalities of Müllerian and renal development and involvement of the cervicothoracic vertebrae.13
Most patients diagnosed with MRKH syndrome present with vaginal agenesis with absent or hypoplastic uterus (96%), renal agenesis/ectopia (88%), vertebral anomalies between C5 and T1 (80%), including fused vertebrae and hemivertebrae. , and short stature has also been described (60%).5 Malformations that include a double kidney have also been described, and within skeletal anomalies, scoliosis and hip dysplasia,14 characteristics that are present in the case presented. MRKH syndrome may be associated with hearing defects, and in rare cases, cardiac (atrial septal defect) and digital anomalies such as syndactyly, polydactyly, or ectrodactyly may occur.9
Other clinical features of MRKH syndrome include short vagina, which can lead to dyspareunia if penetrative vaginal intercourse is attempted, cyclical abdominal or pelvic pain, and uterine factor infertility. The main challenges in the clinical care of these patients involve addressing reproduction, as well as the ability to have penetrative vaginal intercourse.15
DEVELOPMENT OF MESODERMAL TISSUES
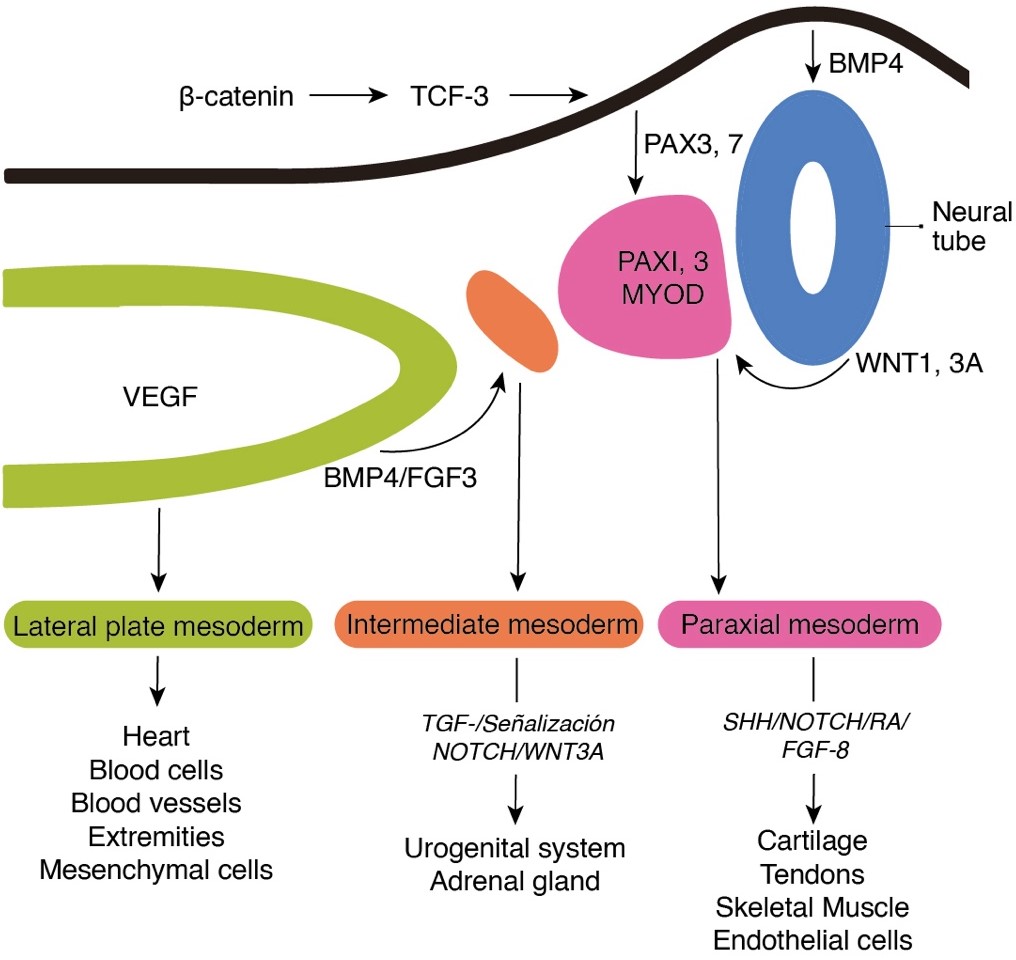
Tissues affected by MRKH syndrome share a common embryonic origin and genetic programs. Proper mesoderm development is critical, as the reproductive tract, kidneys, skeleton, and heart, the organs most commonly affected by MRKH, originate from this germ layer. It has been suggested that the early differentiation of these organs is regulated mainly by the WNT gene,16 bone morphogenetic proteins (BMPs),17 and fibroblast growth factor (FGF).18 During gastrulation, activation of the Tcf-3 gene by β-catenin signaling is a key event that activates specific pathways that drive mesoderm differentiation into paraxial, intermediate, and lateral mesoderm.19
The female reproductive system in humans is derived from the Müllerian or paramesonephric (MD) ducts, which give rise to the uterus, cervix, and upper two-thirds of the vagina around the fifth to sixth week of gestation.20 The mesoderm Paraxial (PM) gives rise to muscles and most of the skeleton. The entire urogenital system, including the reproductive tract and kidneys, is derived from the intermediate mesoderm (IM). Finally, the lateral mesoderm (LM) differentiates into the heart, vascular system, smooth muscles, and limb skeleton (FIGURE 1).21 Like all other embryonic development processes, the development of the Müllerian ducts It is regulated by a coordinated and sequential interaction of molecular pathway networks (MPNs). MPNs consist of transcription factors that regulate the expression of regulatory and effector proteins in carefully directed developmental programming.22

DEVELOPMENT OF THE NEPHRO-URINARY SYSTEM
The first event during the development of the genitourinary tract is the formation of a ductal system that forms the primordium of the future urinary system. This begins in humans with the appearance of the pronephric duct at the end of the third week of gestation. These ducts migrate caudally to form the Wolffian ducts (WD) in humans around week 4 of gestation.24 The development of the Wolffian ducts is a necessary event for the differentiation of the female reproductive tract. The Wolffian ducts form mesonephric tubules in the adjacent mesonephric mesenchyme. In several species of mammals, these tubules perform the functions of an embryonic kidney. In humans, this occurs for only a few weeks before the caudal part of the Wolffian ducts gives rise to the ureteric bud (UB), which invades the surrounding mesenchyme and forms the metanephro, the future permanent kidneys.25
The WNT/β-catenin pathway is critical in Wolffian duct development and is required to maintain Wolffian duct epithelium in a precursor state. The WNT4 gene is expressed in the metanephric mesenchyme and acts as an inducer of mesenchymal to epithelial transmission necessary for kidney development.26
DEVELOPMENT OF THE FEMALE REPRODUCTIVE SYSTEM
After the development of Wolffian ducts, Müllerian ducts (MD) form in a process characterized by three main but poorly understood phases.27 The first begins with the activation of BMP signaling and the induction of the expression of transcription factors. Pax2 and Pax8 in the cranial epithelium adjacent to the Wolffian ducts28 The BMP/PAX2 axis, together with FGF signaling, activates the expression of the transcription factor LXH1 in the coelomic epithelium, stimulating the specification of Müllerian duct epithelial cells29 In the second phase corresponding to invasion, the expression of WNT4 from the mesonephros or coelomic epithelium induces the invagination of these specified cells. Elongation is the third phase regulated by inductive factors from the Wolffian ducts, including WNT4 and WNT9B.30
Tissues affected by MRKH syndrome share common genetic and embryonic programs. Furthermore, early differentiation of these organs is primarily regulated by the same major pathways,16 bone morphogenetic proteins (BMPs),17 and fibroblast growth factor (FGF).18
Both deletion and overexpression of a stabilized form of β-catenin result in urogenital anomalies ranging from renal hypoplasia to agenesis.22 Expression of Pax2 and Pax8 ensures renal lineage specification and survival25; PAX2 induces expression of critical transcription factors, including Lxh1, which is required for Wolffian duct elongation and formation of tubular structures in the developing kidney,30 and the EMX2 gene, which regulates successful renal morphogenesis.31
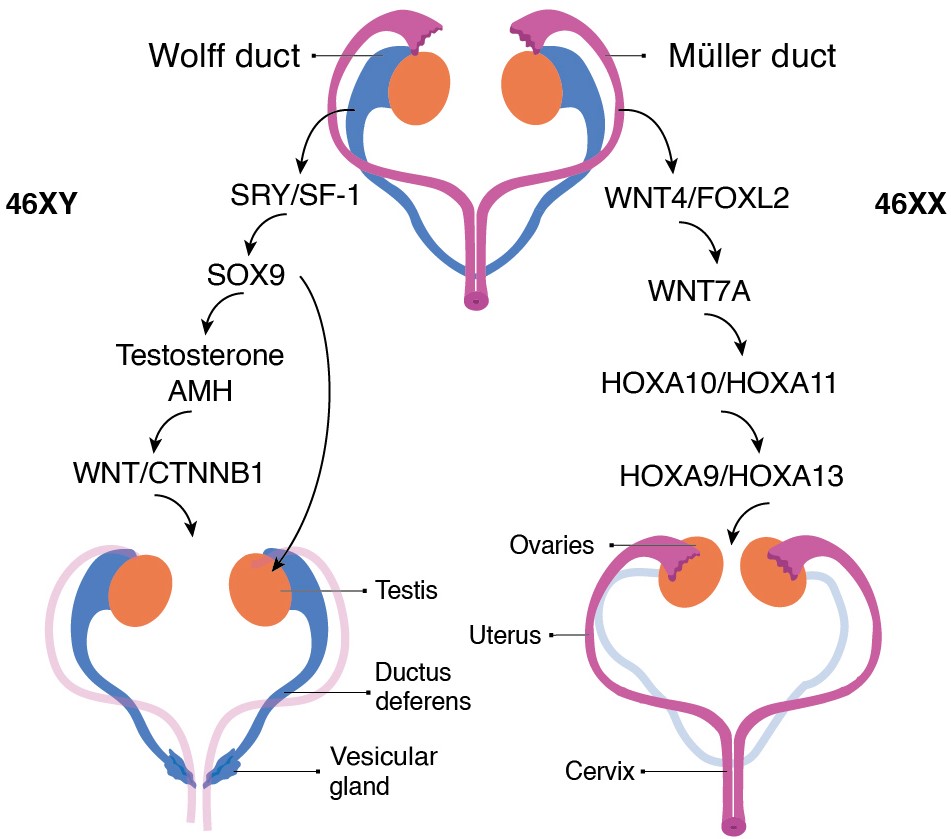
The initiation and invagination of the Müllerian ducts appears to be independent of the Wolffian ducts. However, Wolffian ducts are necessary for Müllerian duct elongation.32 Further differentiation of Wolffian and Müllerian ducts into sex-specific reproductive tracts depends on gonadal development (FIGURE 2). In the male, the SRY gene on the Y chromosome triggers a signaling cascade that leads to the development of testes, which produce testosterone, stimulating the differentiation of Wolffian ducts in the male reproductive tract, and anti-Müllerian hormone (AMH), which causes degeneration of the Müllerian ducts.33 In females, the absence of SRY results in the development of the ovaries through the action of specific genes, including Foxl2 and WNT4. Lack of testosterone and AMH causes regression of the Wolffian ducts and increased differentiation of the Müllerian ducts in the female reproductive tract.34

The anterior regions of the Müllerian ducts develop into the oviducts and uterus. At the same time, the caudal portions fuse in the urogenital sinus to form the uterovaginal duct, giving rise to the cervix and upper vagina.35
GENETIC ETIOLOGY OF MRKH SYNDROME
An alteration of the different pathways involved in the association of developmental abnormalities of the Müllerian ducts with defects in other organs is suggested, not only in sex differentiation but also in the embryonic development of structures derived from the intermediate mesoderm. This syndrome is characterized by a heterogeneous etiology and a pattern of inherence. Sporadic and familial cases have been reported; studies in familial cases support the existence of hereditary predisposition. The results of the most studied familial cases seem to comprise an autosomal dominant mode of transmission with sex-limited expression (female) and incomplete penetration; however, no clear pattern of inheritance has been identified, mainly due to heterogeneity of inheritance pattern and incomplete penetration.36
The identification of germline genetic alterations in a few patients, as described regarding discordant twins for the MRKH phenotype, supports the sporadic nature of the disorder and the lack of heritability. In fact, epigenetic or environmental factors could be the basis for the spontaneous appearance of congenital uterine malformations.37
This etiological diversity of MRKH is also evidenced by the occurrence of multiple genetic and genomic lesions in affected patients. The genes that cause MRKH syndrome have been identified according to their location within the genomic rearrangements observed in patients. Many other candidates belong to genetic pathways that drive embryonic development or have emerged from studies of animal models or syndromic conditions in which uterine malformations are a frequent clinical manifestation. A genotype-phenotype correlation for WNT4 mutations has been reported in MRKH patients with hyperandrogenism, suggesting a clinically and genetically distinct MRKH subtype associated with this gene, likely involved in androgen regulation.38
Several candidate genes have been proposed for genetic analyses of women affected by MRKH syndrome. A few are mentioned in TABLE 2, whose fundamental role in the development of the urogenital tract has been established.
Among the genes associated with the development of MRKH syndrome is the estrogen growth regulation gene in the androgen-regulated breast cancer type 1 gene (GREB1L), which is a coactivator of the retinoic acid receptor (RAR) gene, whose activation regulates the retinoic acid pathway,54 and which has been shown to trigger the differentiation of Müllerian epithelial cells and establish the boundary between the uterus and vagina55; therefore, GREB1L plays an indispensable role in the development of embryonic metanephrosis and the genital tract.56
Many patients diagnosed with Rokitansky syndrome manifest the MURCS association because the pattern of pathophysiological development is similar in both conditions; therefore, patients with Rokitansky syndrome should be studied to detect the characteristic alterations of the MURCS association and avoid underdiagnosis.57
MANAGEMENT OF PATIENTS WITH DIFFERENT MÜLLERIAN ANOMALIES
Müllerian anomalies are associated with high rates of preterm delivery, premature rupture of membranes, fetal malpresentation, and perinatal mortality.58 There is insufficient evidence to suggest that Müllerian anomalies are associated with infertility, as similar rates have been reported compared to the general population.59 However, in patients diagnosed with MRKH syndrome, specifically MURCS association, uterine surrogacy, and more recently the implementation of uterine transplantation, seem to be the only feasible options for patients with this pathology. However, the latter practice is widely debated.
Depending on the type of presentation diagnosed and the severity of the findings, part of the treatment of patients with Mayer-Rokitansky-Küster-Hauser syndrome includes psychological counseling for them and their families since this diagnosis has repercussions on future reproduction.60 The treatment is mainly aimed at creating a neovagina that allows sexual intercourse. Surgical and non-surgical treatments are available. Surgical options include graft placement using the McIndoe61 or Vecchietti62 procedure. The mechanical approach includes the application of dilators, as described in Frank’s procedure.63
Differential diagnoses include androgen insensitivity, the presence of a transverse vaginal septum, isolated distal vaginal agenesis, and imperforate hymen.64
CONCLUSIONS
Müllerian anomalies are a set of abnormalities of sexual development during the embryonic period. Within this group of abnormalities is MRKH syndrome, in which it is suggested that it is a defect of mesodermal origin. The genetic etiology of MRKH syndrome is not clear, nor is it one hundred percent established.
Some authors agree on separating the MRKH syndrome from the MURCS association, and others consider the MURCS association as a phenotypic variant of the MRKH syndrome when abnormalities in other organs accompany it. The first clinical feature is usually primary amenorrhea. The main challenges in the clinical care of these patients involve addressing reproduction as well as the ability to have penetrative vaginal intercourse. As for the fertility of people with MRKH, surrogacy is an option.
Although this syndrome is not uncommon, subtype 2 of MRKH syndrome is, so the disclosure of a complete clinical case is important when trying to understand and address it. However, until the cause of these clinical entities is established, it will be difficult to deny or affirm whether the atypical forms of MRKH syndrome and the MURCS association belong to the same phenotypic spectrum originating from common or related causes or are simply different entities.
Authors’ Contribution - CRediT
Investigation: Julio César Rodríguez Verduzco (Lead). Project administration: Julio César Rodríguez Verduzco (Lead). Supervision: Julio César Rodríguez Verduzco (Lead). Writing – original draft: Julio César Rodríguez Verduzco (Lead). Data curation: José Ines González Tapia (Lead). Validation: José Ines González Tapia (Lead). Formal Analysis: Nelly Ivette Martinez Galindo (Lead). Funding acquisition: Alexis Eliseo Santos Rodríguez. Methodology: Ana Norma Gricelda Becerril González (Lead). Conceptualization: Fernando Mancilla Hernández (Lead). Resources: Fernando Mancilla Hernández (Lead). Writing – review & editing: Fernando Mancilla Hernández (Lead). Software: Martha Camila Correa Castillo (Lead). Visualization: Yaser Laurel Lujan (Lead).