
Background
A well-executed cycle stimulation that optimizes the oocyte yield correlates with the success of in vitro fertilization (IVF).1,2 Urinary and recombinant gonadotropins are used in a personalized way with age, ovarian reserve markers, body mass index (BMI) and response to previous stimulation considered to achieve the stimulation goals.3 Based on these parameters, patients are often identified as expected normal, hyper or poor responders.
With respect to poor responders, various treatment protocols have been evaluated to optimize oocyte yield.4 Most of these protocols involve the use of increased dose gonadotropins, which is based on outcomes of gonadotropin dose finding studies where more follicles and oocytes are obtained as the gonadotropin dose is increased.5 However, this practice fails to follow the same trend in the number of embryos and good quality blastocysts.2,5
Recent ESHRE guidelines question the benefit of gonadotropin doses exceeding 300 IU daily,3 yet clinical practice often employs doses of 450-600 IU in poor responders, highlighting the disconnect between evidence and practice. Several randomized controlled trials (RCT) have compared clinical outcomes using a lower versus a higher dose of gonadotropin in poor responder patients6–13 but have a number of inconsistencies, including the varying criteria to identify poor responders, the use of different stimulation protocols (antagonist vs long protocols), the use of oral agents where international unit conversion is not possible, dose escalation during cycles, and inconsistent primary endpoints, making meta-analysis and clinical application challenging.14 More recently, POSEIDON and Bologna criteria have emerged as standard tools to identify poor responders.15,16 Beyond clinical outcomes, the economic implications of high-dose protocols are substantial, with gonadotropin costs representing a significant portion of IVF treatment expenses, particularly relevant for patients who may require multiple cycles.
Therefore, the aim of our RCT was to compare clinical outcomes, including live birth rate (LBR) and cumulative (c)LBR, using a lower versus a higher dose gonadotropin regimen in expected poor responders by POSEIDON criteria.
Methods
This multicenter RCT took place in six IVF centers in Hungary. IRB approval was obtained prior to study start (52007-16/2021/EÜIG) and the study was conducted in accordance with the principles set forth in the Helsinki Declaration and local regulations. The trial was registered at Clinical Trials (NCT05103228; October 21, 2021). All study sites were required to obtain an additional insurance policy to cover the clinical trial as per local regulations. Recruitment was initiated in December 2021 with complete recruitment expected within one year. Due to slow enrollment, the recruitment period was eventually extended until July 2023 at which point the study was closed out due to lack of funding for insurance extension. Frozen embryo transfers resulting from the fresh cycles were included up until the end of 2023 and pregnancy outcome data was collected up until September 2024. All participants were given a detailed explanation of the study goals, methods and potential disadvantages, and were required to sign an informed consent prior to enrollment.
Inclusion-Exclusion
The inclusion-exclusion criteria are shown in Table 1. None of the cycles involved pre-implantation genetic testing (PGT) for aneuploidy as it is not allowed by Hungarian law.
Randomization
Enrollment into the study was performed prior to gonadotropin stimulation. Eligible participants were randomly assigned to their treatment protocol based on an online randomization list that was generated prior to the start of the trial (www.randomizer.org). Patients were randomized to either a lower vs. higher dose of gonadotropins and to Follitropin-α or δ use. Follitropin-α and δ have been shown to result in similar clinical outcomes and comparable doses of follitropins (150 IU FSH-α or the equivalent dose of 10 μg FSH-δ) were used in the lower (LD) and higher (HD) dose groups.17,18 Randomization was performed by the local principal investigator (PI) once consent was obtained. Only the PI had access to the randomization list and provided the treatment allocation to his/her colleagues once the patient has signed consent and was ready to enter the study. Patients were only enrolled once regardless of their treatment outcome. Neither patients nor the providing physician were blinded to the treatment assignment.
Sample size
To determine the sample size needed, we assumed a 20% clinical pregnancy rate (CPR) in our patient population that fit the inclusion criteria based on the results provided by the participating clinics. Given that we expected a greater oocyte yield in the higher dose group with greater available embryos and the potential for more embryo transfers. As such, we anticipated a 50% increase in cumulative (c)CPR and with an expected 20% dropout rate for low response and cycle cancellation, 350 participants per study arm were needed to achieve a power of 80% and an alpha error of 5%, to show superior results with the high dose FSH treatment.
Due to recruitment challenges and early study termination, the final enrollment of 190 patients was substantially below our target of 750 participants. This severe underpowering transformed our study from a definitive efficacy trial into an exploratory analysis.
Treatment
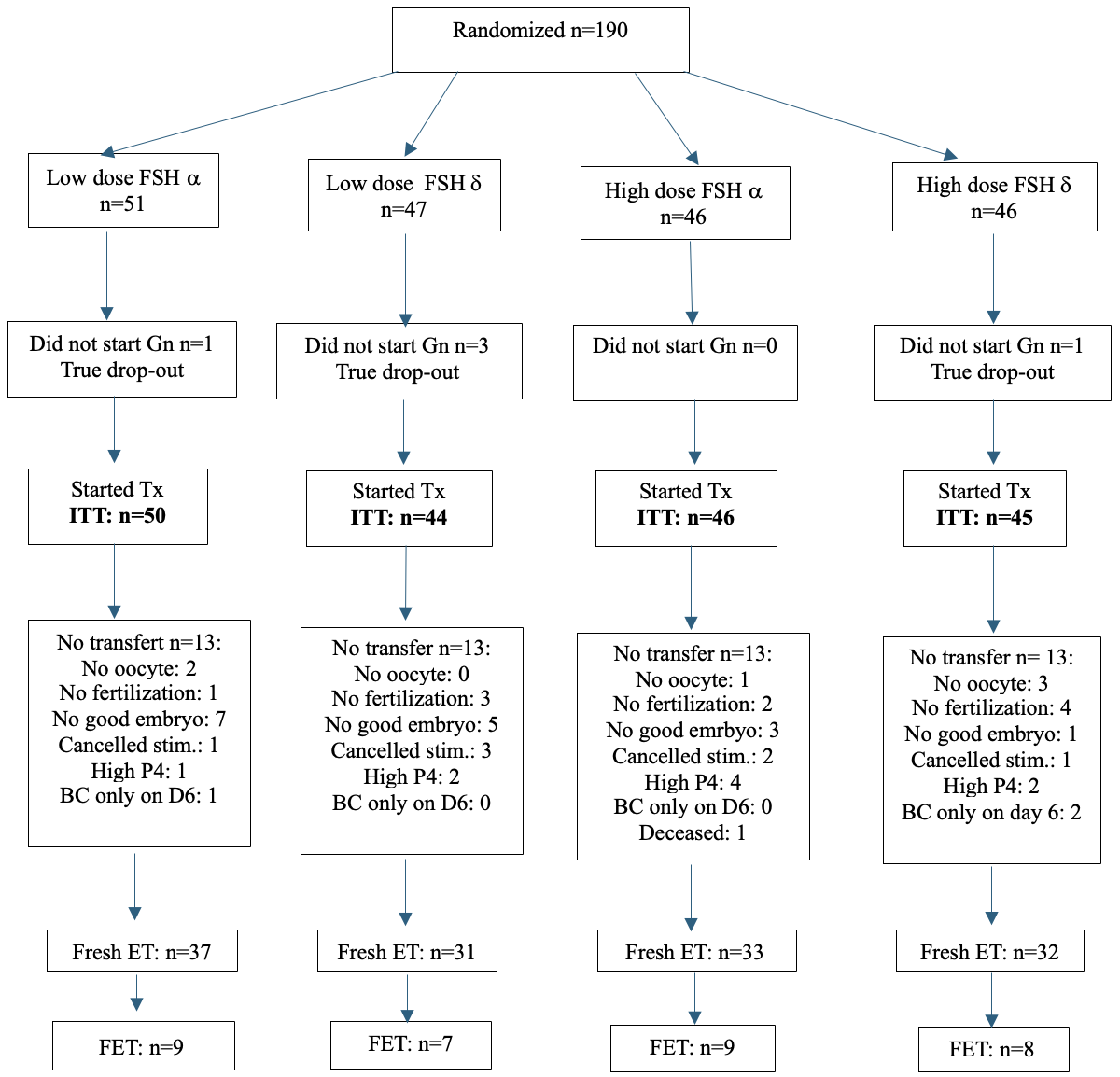
Patients were randomly assigned to a lower (225 IU/day; 150 IU FSH + 75 IU hpHMG) or a higher dose (375 IU/day, 225 IU FSH + 150 IU hpHMG) gonadotropin combination and the use of Follitropin-α or δ. Stimulation was started on day 2 or 3 of a spontaneous cycle or on the 5th day after contraceptive pill use. Both follitropin-α (FSH-α: Gonal-F, Merck Serono; Ovaleap, Theramex Ireland Ltd; Bemfola, Richter Gedeon Nyrt.) and follitropin-δ (FSH-δ: Ferring Pharmaceuticals) were used in combination with highly purified human menopausal gonadotropin (hpHMG: Meriofert, IBSA Farmaceutici Italia Srl). (Figure 1) The drug dose had to be maintained throughout the study unless there was evidence of hyper-response (estradiol [E2] level >4000 pmol/l on day 6 of stimulation or >15 follicles over 10 mm on any day of stimulation) in which case a dose reduction or cycle cancellation could be considered.
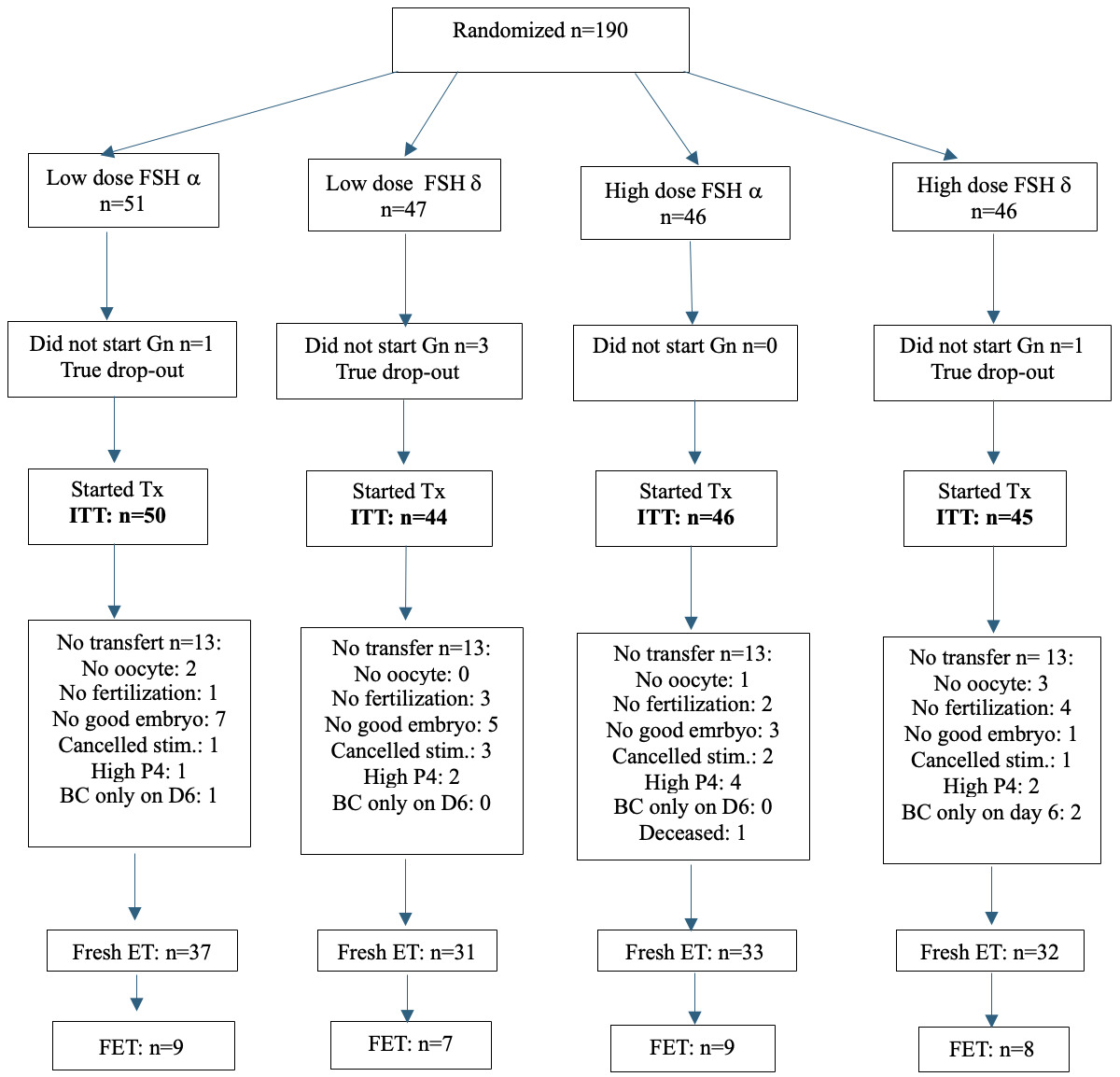
Vaginal ultrasound and serum hormone measurements were used to monitor response to stimulation from day 5-6 of stimulation and every 2-3 days thereafter. Gonadotropin releasing hormone antagonist (GnRH-ant, Cetrotide, Merck-Serono, Ganirelix, Richter Gedeon Nyrt.) was started once the lead follicle reached 12-14 mm in diameter. When the largest follicle reached >17 mm, human chorionic gonadotropin (hCG; Ovitrelle, Merck-Serono) or in the case of hyper-response GnRH agonist (0.2 mg Gonapeptyl, Ferring Pharmaceuticals Ltd) trigger was given, followed by the transvaginal, ultrasound-guided retrieval 35-36 hours later. Cycles in which there was no response to stimulation (no follicles over 10 mm after 10 days of stimulation) were cancelled. Retrieved oocytes were fertilized by conventional insemination or ICSI fertilization. In cycles with a preovulatory progesterone rise (progesterone at last scan >1.5 ng/ml), elective cryopreservation was recommended. One or two embryos were transferred (ET) transcervically under ultrasound guidance using soft catheters (Wallace, Smith Medical International Ltd., UK) after 3-5 days of group culture. The number of embryos transferred was based on local protocols and an agreement between the couple and the physician. Surplus good quality embryos were vitrified.
The luteal phase was supported with vaginal (Utrogestan, Lab Besins International SA or Cyclogest, Richter Gedeon Nyrt.) or subcutaneous (s.c., Prolutex, IBSA Farmaceutici Italia Srl) progesterone starting the day after the retrieval. Pregnancy was confirmed by serum β-hCG 12-14 days following embryo transfer and clinical pregnancy was assessed by ultrasound 2-3 weeks after a positive test.
Frozen embryo transfers (FET) were preferably performed in a natural cycle using hCG trigger. Some FETs (based on a discussion between the provider and the patient) were carried out in a hormone replacement cycle using oral estradiol (Estrofem Novo Nordisk AS) and vaginal and/or subcutaneous progesterone added when endometrial thickness reached >7 mm.
Outcome parameters/ Data collection
Data including baseline parameters, stimulation, embryology and clinical outcome parameters were collected in data collections sheets prepared in advance at each site and were then compiled into one final dataset as shown in Table 2. The primary outcome of interest was cCPR. As secondary outcomes, LBR and cLBR with lower vs higher dose and comparison of different follitropins were also studied. CP was defined as an intrauterine sac at 6-8 weeks gestation, LB as the birth of a singleton or twin after 24 weeks of gestation, and cumulative (c) LB as a LB following the fresh or any of the frozen ETs resulting from the same retrieval.
Statistics
After enrollment of the first 350 patients, an interim analysis was planned to assess sample size assumptions and to test for the effect of gonadotropin dose on outcomes. However, due to various issues including patient death, medication shortages, and insurance requirements, recruitment was stopped, and analysis was performed on 190 enrolled participants, representing 25% of the planned sample size. Post-hoc power analysis revealed that with 190 patients, our study had approximately 15% power to detect the originally anticipated 50% increase in cumulative clinical pregnancy rate and insufficient power (<20%) to detect smaller but clinically meaningful differences of 15-25%. This severe underpowering transformed our study from a definitive efficacy trial to an exploratory analysis.
Continuous variables were summarized as means ± standard deviations (SD), while categorical variables were presented as counts (n) and percentages (%). Data normality was assessed using Shapiro-Wilk tests and QQ-plots. Variables showing significant departure from normality (total gonadotropin dose, stimulation duration, and number of oocytes retrieved) were log-transformed based on Akaike Information Criterion (AIC) model comparison and residual assessment.
For the 2×2 factorial design, we tested main effects of gonadotropin dose (225 IU vs 375 IU daily), FSH type (follitropin-α vs follitropin-δ), and their interaction. Linear mixed-effects models were employed for continuous outcomes with gonadotropin dose group and FSH type as fixed effects and participating center as a random intercept. Generalized linear mixed-effects models with logit link function were used for binary outcomes using the same fixed and random effects structure. Model assumptions were verified through residual plots and QQ-plots. When model assumptions were violated, non-parametric tests (Wilcoxon rank-sum for continuous variables, Fisher’s exact tests for categorical variables) were employed. Missing data were handled using complete case analysis for the primary analysis, with multiple imputation sensitivity analyses performed for outcomes with >10% missing data. Given the exploratory nature of this severely underpowered study, no adjustment for multiple comparisons was applied, but all p-values should be interpreted as exploratory findings with increased Type I error risk.
Statistical significance was set at p < 0.05, though emphasis was placed on effect sizes and confidence intervals given the underpowered design. Analysis was performed as both intention-to-treat (including all 190 randomized patients) and as treated (including only the 133 patients who underwent embryo transfer). All analyses were conducted using R statistical software with appropriate packages for mixed-effects modeling and multiple imputation.
Results
Of the planned 750 enrolled subjects, 190 were randomized (Figure 1). Fresh cycle embryo transfer took place in 133 patients: 68 in the lower dose FSH, while 65 in the higher dose FSH arms. (Figure 1).
This did not represent the interim analysis that was initially planned. Rather, given the number of issues that impacted recruitment during the study including a death of a patient that was considered unrelated to her treatment (see below); a shortage of GnRH-ant that occurred twice for two months; and the mandated requirement for additional insurance after the estimated study date completion, the joint decision of investigators was to close the study.
Intent-to-Treat and As Treated Analysis
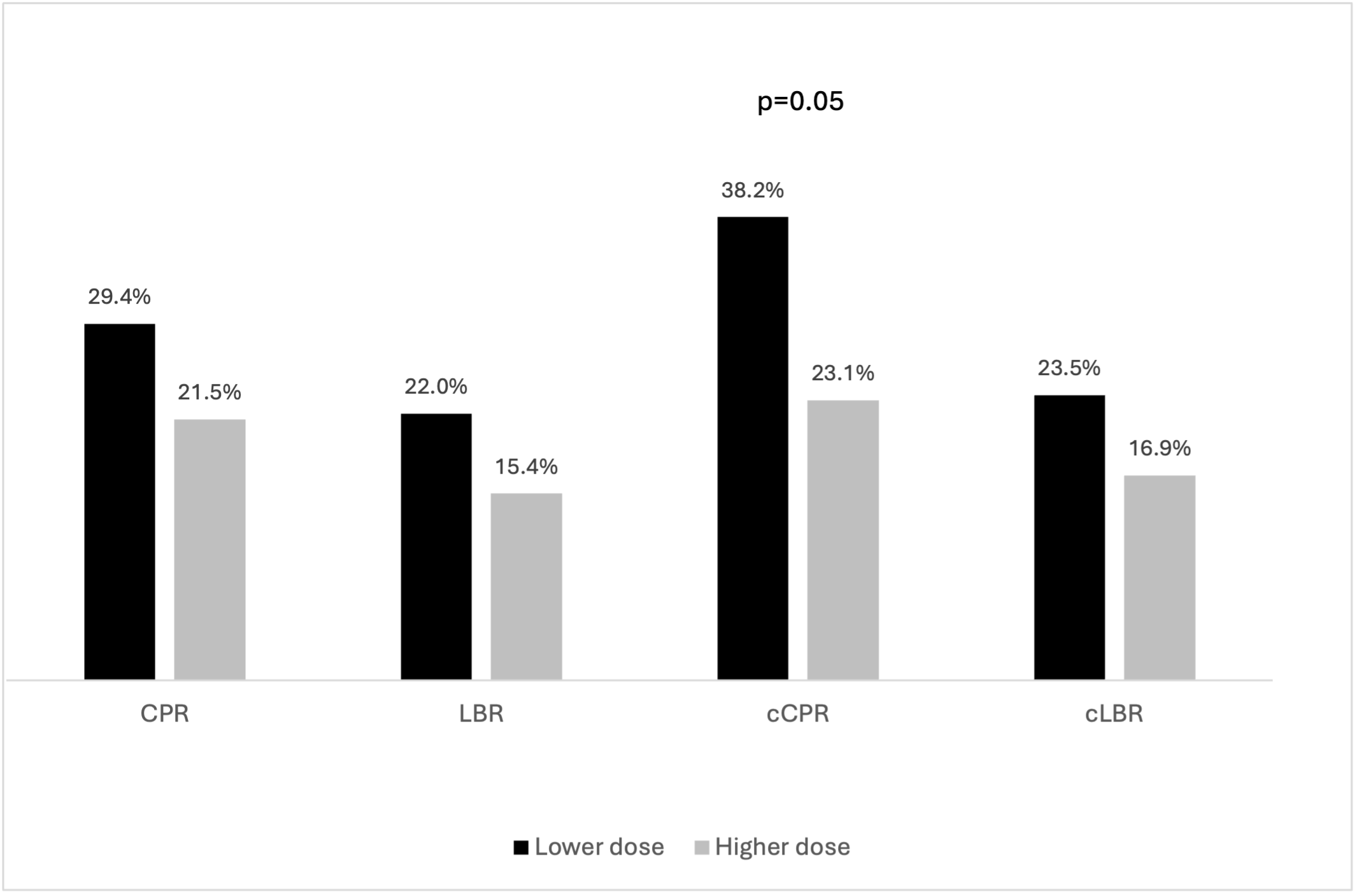
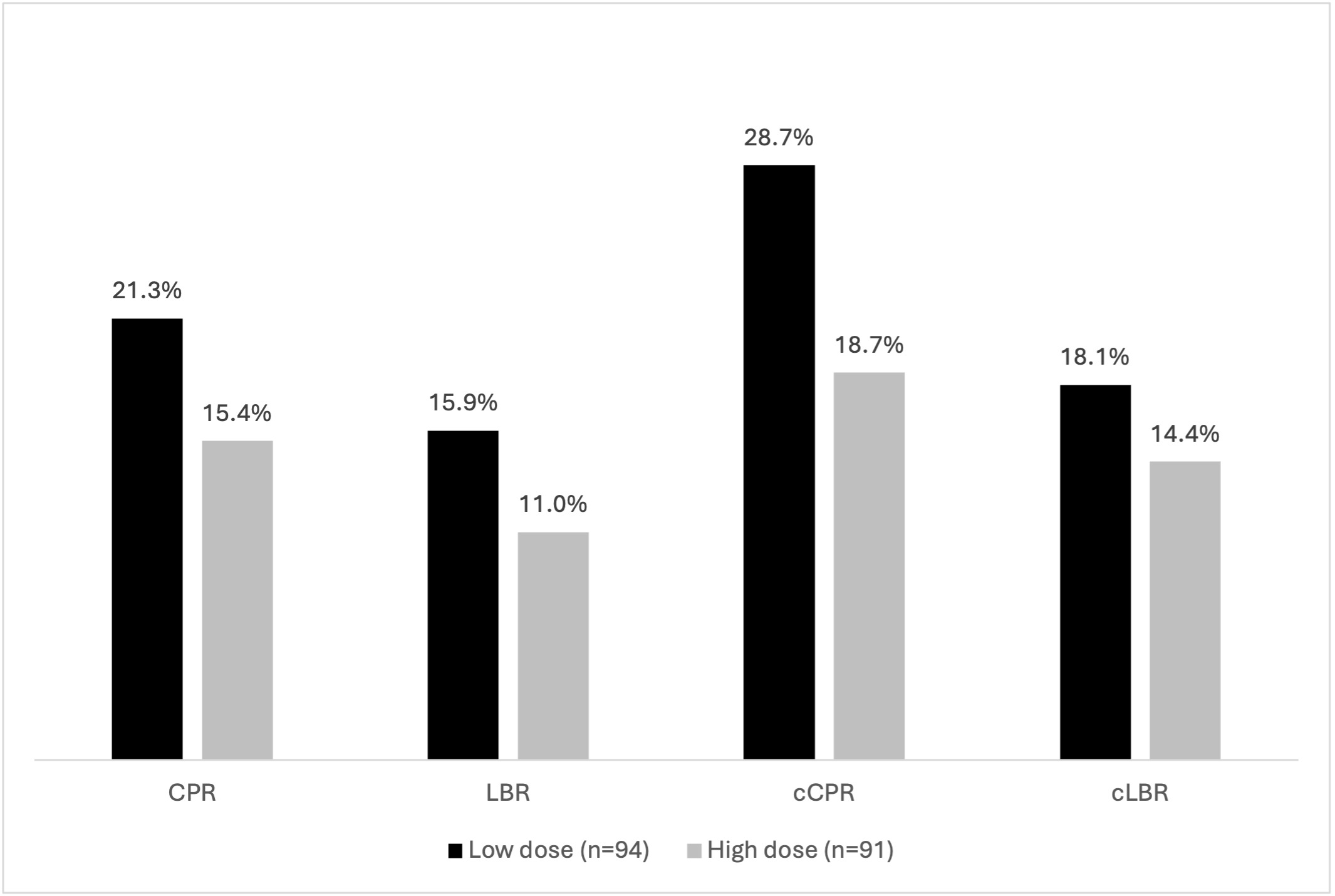
Lower and higher dose FSH groups were well-balanced regarding baseline characteristics. (Table 3). With respect to cycle stimulation characteristics and embryology outcomes, significantly less gonadotropins were used in the lower dose FSH arm (2189 IU vs. 3550 IU, p<0.001). (Table 4) Regarding the ITT analysis, no differences were seen with respect to clinical outcomes (RR CPR: 1.07;0.93-1.23; RR LBR: 1.05;0.94-1.18; RR cCPR: 1.14;0.97-1.34; RR cLBR: 1.04;0.92-1.18) (Figure 2). In the as treated analysis, while cCPR was significantly higher in the lower dose arm (38.2% vs 23.1%, p=0.05), significance was not seen for cLBR (RR CPR: 1.11;0.91-1.35; RR LBR: 1.08;0.92-1.27; RR cCPR: 1.24;0.99-1.56; RR cLBR: 1.08;0.91-1.29). (Figure 3)
_low_versus_high_dose_o.png)
Comparison of Follitropin-α and δ
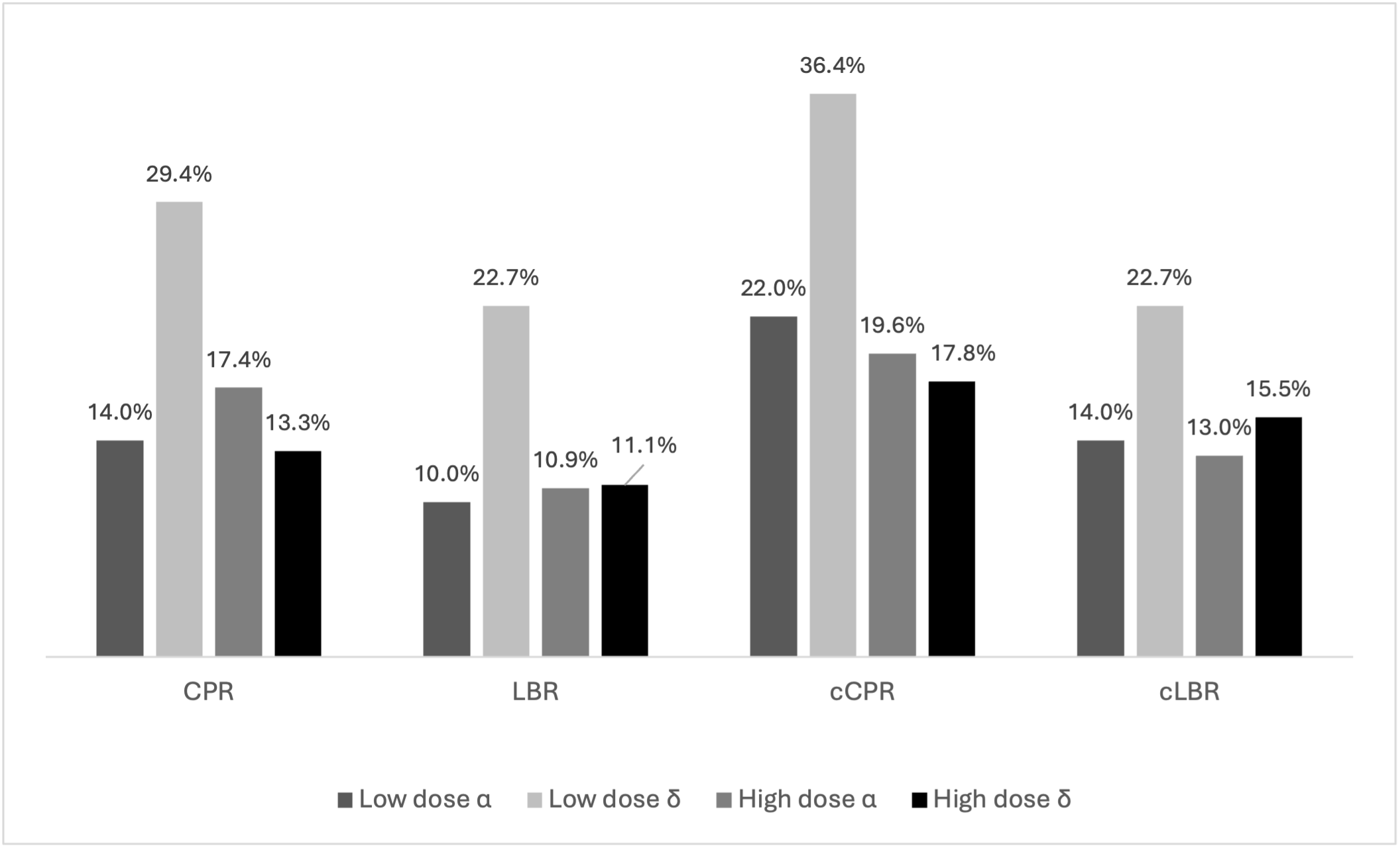
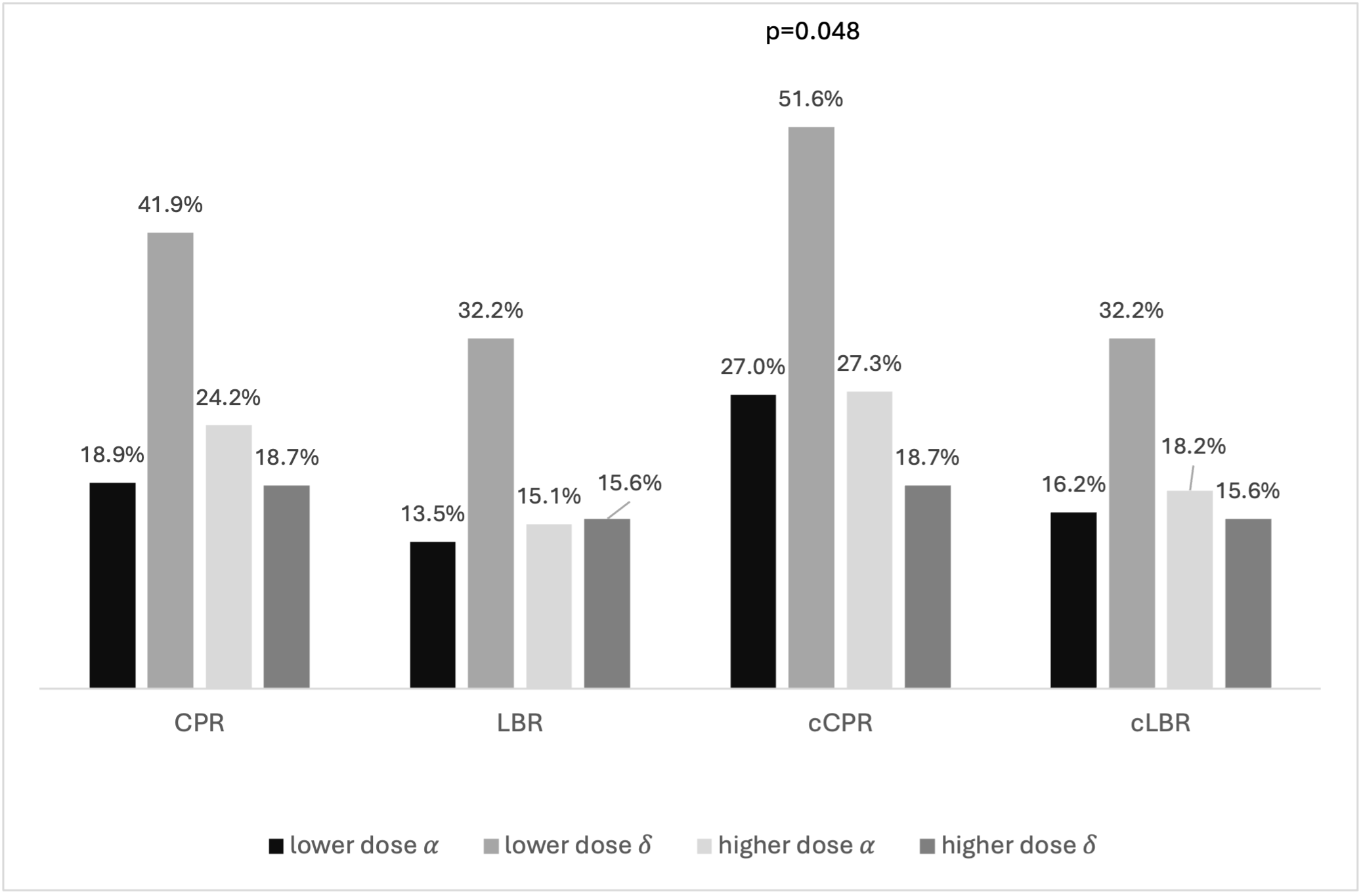
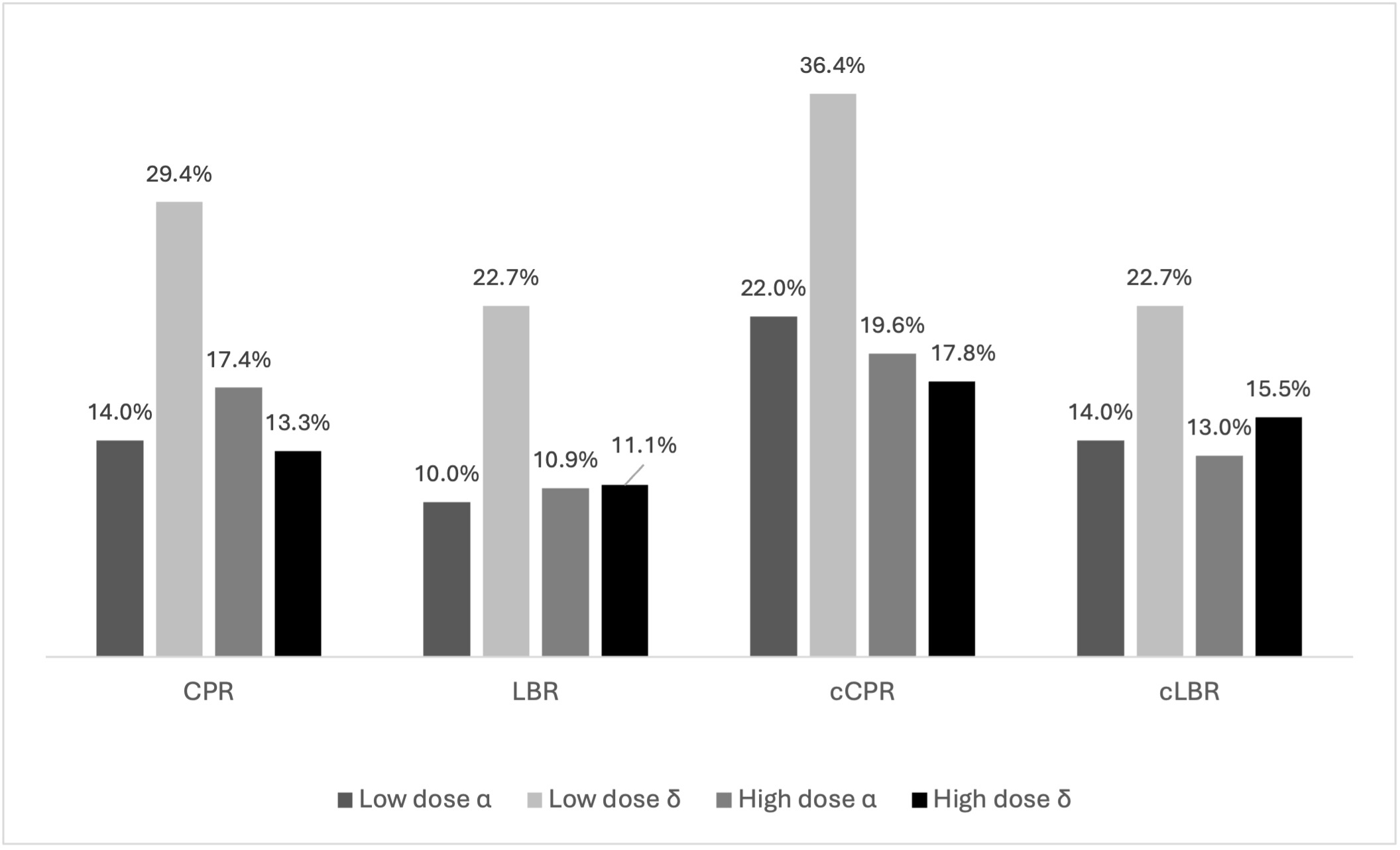
When outcomes were compared using different follitropin preparations (FSH-α vs δ), demographic characteristics, cycle stimulation, embryology parameters (Supplemental Table 1), and clinical outcomes (Figure 4) were comparable in the four subgroups. Regarding analysis for as treated cycles, while cCPR significantly differed (LD α: 27.0%, LD δ: 51.6%, HD α: 27.3%, HD δ:18,7%, p=0.048), this was not noted for cLBR. (Figure 4, 5)
_based_on_and_follitropin_type.png)
Adverse events
One patient died after randomization in the increased dose FSH group while undergoing ovarian stimulation. Her death after the coroner’s exam, revealed she suffered from an autoimmune disease that she failed to disclose and flared leading to multiorgan failure and eventually her death that was considered unrelated to her fertility treatment. No other severe adverse events (complications during the retrieval procedure requiring intervention/ hospitalization, ovarian hyperstimulation syndrome or thromboembolic event) were observed during the study.
Discussion
Our results do not support superior stimulation, embryology or clinical outcomes when higher dose gonadotropins are used among expected poor responders during IVF. Furthermore, other than cCPR, different follitropin subtypes do not result in improved outcomes.
Induction of multi-follicular development is a critical step of IVF as with each step there is some expected loss progressing from follicle aspiration to embryo implantation.19 Dose-finding studies of gonadotropins have shown that with higher gonadotropin doses, more follicles are recruited and greater number of oocytes collected5 but not paralleled by an increase in blastocysts.2,5 This “contradiction” suggests that the quality of oocytes in a stimulated cohort is heterogeneous and over certain number of oocytes no further clinical gain can be expected.
A wide variety of treatment options have been explored in this group of patients4 including the use of higher doses of gonadotropins.6–13 Nonetheless, studies are inconsistent including varying stimulation protocols,6,7,9,12 differing gonadotropin types with dose increases leading to an eventual “high-dose” administration in the low dose arms,8 and outcome parameters tested have varied (CPR, ongoing PR, LBR) with the most important outcome, cLBR reported only in a small subset of studies.12,13 Finally, varying criteria to define poor responders10,12,13 makes it at best difficult to compare the results of the various studies.14
Nonetheless, despite these study design differences; studies have overall been “homogenous” including lower cancellation rate12 and greater retrieved oocytes with higher dosing.10,12 Moreover, others have suggested an adverse effect of high dose FSH on oocyte/ embryo quality,20–23 though not a consistent finding.24–26 Additionally, no differences in clinical outcome, including LBR and cLBR have been demonstrated, resulting in ESHRE COS guideline questioning the benefit of a gonadotropin dose in excess of 150 IU daily and further advising against the use of a dose higher than 300 IU daily.3
Nonetheless, given the diagnostic, treatment and reporting inconsistencies of previous studies, we attempted our RCT using the 2016 POSEIDON classification that was introduced to define a more universal identification of poor responders.16 To avoid previous inconsistencies, we maintained a fixed drug dosing in both the lower and increased dose arms. However, despite this standardized approach, we failed to identify a benefit with either lower or higher dose gonadotropins and FSH formulations.
Our study has several strengths. We used standard definitions to identify expected poor responders; patients were randomly assigned to a fixed lower or higher drug dose without dose-adjustment and tested clinically relevant outcomes including cLBR as a measure of the full reproductive potential of the IVF cycle. Further, the multi-center design improves the external validity of the findings though all clinics belong a single health care system following national regulations, and our analysis was performed as an intent-to-treat and as treated.
This study also has several important limitations that significantly affect the interpretation of our findings. The most critical limitation is the severe underpowering resulting from premature study termination, with only 190 participants enrolled (25%) of the planned 750-patient sample size. This dramatic shortfall fundamentally transforms our investigation from a definitive efficacy trial to an exploratory pilot study. For the observed difference in our primary outcome (28.7% vs 18.7% cCPR), approximately 277 participants per group would have been required to achieve 80% statistical power This severe underpowering has profound implications for result interpretation. Our negative findings should not be interpreted as definitive evidence that higher gonadotropin doses provide no benefit to poor responders. Rather, they represent insufficient statistical power to detect potentially clinically meaningful differences, creating substantial risk of Type II error. Thus, while our results are consistent with no benefit from moderate dose escalation and support current evidence-based guidelines, they cannot definitively exclude the possibility that dose increases provide meaningful clinical improvements.
Several methodological limitations further compromise our findings. The open-label design introduced potential performance and detection bias, as neither patients nor providers were blinded to treatment assignments. This could have influenced treatment decisions, monitoring intensity, and outcome assessment. The unplanned early termination due to logistical issues (patient death unrelated to treatment, medication shortages, insurance requirements) rather than pre-specified efficacy or futility criteria reduces confidence in our negative results and prevented the planned interim analysis that might have informed sample size adjustments. Our study’s generalizability is also limited by several factors. All participating centers were located within a single healthcare system in Hungary, potentially limiting applicability to other healthcare settings with different patient populations, protocols, or resource availability. The exclusion of preimplantation genetic testing and other advanced reproductive technologies due to local regulatory restrictions limits relevance to centers where these techniques are standard practice. Additionally, our definition of “high dose” (375 IU daily) represents a moderate increase compared to doses commonly used in clinical practice (450-600 IU), meaning our findings may not apply to truly high-dose protocols. The study design itself presents limitations. Our sample size calculation was based on potentially unrealistic assumptions, including a 50% relative increase in clinical pregnancy rates with higher dosing and a baseline rate that may have been optimistic for this poor-prognosis population. The 2×2 factorial design, while efficient, may have been underpowered to detect meaningful interactions between gonadotropin dose and FSH formulation. The broad inclusion criteria (BMI 18-35 kg/m²) encompassed substantial patient heterogeneity that might have obscured subgroup-specific effects. Data collection and analysis limitations include the lack of standardization of embryo culture conditions and transfer policies across centers, which could have introduced inter-center variability beyond what our statistical models could adequately control. The absence of comprehensive cost-effectiveness analysis, despite mentioning economic implications, limits our ability to provide definitive guidance on the economic aspects of dosing decisions. Furthermore, our follow-up, while adequate for pregnancy outcomes, did not extend to long-term maternal and neonatal health outcomes. Finally, the exclusion of patients with certain baseline characteristics (previous poor response, specific medical conditions) may limit the applicability of our findings to the broader population of poor responders encountered in clinical practice. The restriction to antagonist protocols means our results may not apply to other stimulation approaches commonly used in poor responders. Despite these substantial limitations, our study provides valuable exploratory data suggesting that moderate gonadotropin dose increases (from 225 to 375 IU daily) do not improve clinical outcomes in poor responders, while increasing treatment costs. These findings support current evidence-based guidelines recommending against routine high-dose protocols, though definitive conclusions require confirmation in adequately powered studies.
Although we did not collect data on medication expense, using lower dosing and taking average Hungarian medication and treatment expenses into consideration, the approximately 1400 IU less gonadotropin used in the lower dose arm resulted in a 15% overall treatment expense reduction.
In conclusion, our findings suggest that a modest dose increase does not improve outcomes, raising questions about the utility of more aggressive dosing protocols. The administration of a more modest dose is likely to be associated with significant cost savings without compromising outcomes though proper cost-effectiveness analysis has to be part of future trials. Given the noted differences in cCPR, studies to assess the impact of newer gonadotropins including follitropin-δ should be considered in expected poor responder women undergoing IVF. These findings support current evidence-based guidelines recommending against routine high-dose protocols, though definitive conclusions require confirmation in adequately powered studies.
Clinical Trials Registration
NCT05103228 (Oct 21, 2021)
Conflict of interest
None of the authors have any conflict of interest to declare that could inappropriately influence or bias the work.
Funding
The research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgements
None.
Authors Contribution - CRediT
Conceptualization: Peter Kovacs; Steven R Lindheim
Data curation: Peter Kovacs; Janos Zadori; Peter Boga
Formal Analysis: David U Nagy; Emilie Sandfeld; Steven R Lindheim
Investigation: Peter Kovacs; Janos Zadori; Peter Boga
Methodology: Peter Kovacs; David U Nagy
Project administration: Peter Kovacs
Supervision: Peter Kovacs; David U Nagy
Visualization: David U Nagy; Emilie Sandfeld; Steven R Lindheim
Writing – original draft: Peter Kovacs; Steven R Lindheim
Writing – review & editing: Peter Kovacs; David U Nagy; Janos Zadori; Peter Boga; Emilie Sandfeld; Steven R Lindheim